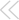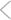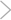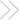1. Pain Makes CaMKII Go Up and Down
Max Larsson and Jonas Broman
Aparticularly intense painful stimulus enhances the sensation evoked by subsequent noxious stimuli. This hyperalgesia may result from facilitation of sensory neurotransmission in the spinal cord, perhaps by mechanisms similar to synaptic plasticity in other pathways. In this week's Journal, Larsson and Broman used immunolabeling of calcium/calmodulin-dependent kinase II (CaMKII) to track the associated modulation of spinal cord pathways. The translocation of autophosphorylated CaMKII to the postsynaptic density makes it one marker of synaptic plasticity. The authors injected rat hindpaws with capsaicin to induce hyperalgesia. Transganglionic tracing of the capsaicin injection site marked the nonpeptidergic class of nociceptors, but not peptidergic nociceptive neurons that express substance P and calcitonin generelated peptide. Phosphorylated CaMKII more than doubled in dorsal horn synapses formed by peptidergic neurons and fell by about one-half in synapses formed by labeled, nonpeptidergic neurons. In low-threshold mechanoreceptor neurons, CaMKII expression did not change with capsaicin treatment.
2. D1, GAD, and Working Memory
Nobuhide Kobori and Pramod K. Dash
Kobori and Dash examined deficits in working memory after a mild concussive brain injury that did not cause overt neuronal loss. The authors targeted the medial prefrontal cortex (mPFC), known to be important for working memory (WM). Rats performed a delay match-to-place task, in which they escaped a water maze to a hidden platform (the location trial), were removed from the maze, and then relocated the platform (the match trial). Brain-injured rats displayed a random search pattern during the match trial indicative of impaired WM. The memory deficits were associated with neurochemical changes in mPFC, including higher levels of D1 dopamine receptors and more neurons detected by immunolabeling for GAD67, the GABA-synthesizing enzyme. GABA receptor antagonists improved WM, and remarkably a single dose of the D1 receptor antagonist SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1 H-3-benzazepine hydrochloride] reduced GAD67 expression and improved WM for several days. Pass the SCH23390, please.
3. Focusing Attention on the Thalamic Reticular Nucleus
Kerry McAlonan, James Cavanaugh, and Robert H. Wurtz
In this week's Journal, McAlonan et al. explore the "attentional searchlight" hypothesis for the thalamic reticular nucleus (TRN). The TRN consists of a thin layer of GABAergic neurons that receive excitatory inputs from the lateral geniculate nucleus (LGN) and cortex and send inhibitory output back to the LGN. The authors recorded spike rates of individual visual TRN neurons in monkeys. These neurons had a high basal firing rate, providing tonic inhibition to LGN, and short latency (∼25 ms) transient responses to visual stimuli. Because the TRN receives multimodal input, the authors examined responses in which monkeys shifted their attention from auditory to visual stimuli. Spiking of TRN neurons increased with the attentional shift, but latency and duration remained the same. The authors suggest that TRN neurons facilitate visual detection and discrimination by switching visual LGN neurons from tonic to burst firing, or by surround inhibition of nonvisual LGN neurons.
4. SOD1 Mutants in and out of Mitochondria
Daniel Bergemalm, P. Andreas Jonsson, Karin S. Graffmo, Peter M. Andersen, Thomas Br?nstr?, Anna Rehnmark, and Stefan L. Marklund
CuZn superoxide dismutase (SOD1)mutations underlie some cases of familial amyotrophic lateral sclerosis (ALS). Mutations cause a cytoxic gain-of-function of this antioxidant molecule that is expressed in the cytosol as well as the mitochondrial intramembrane space. Transgenic mice expressing high levels of stable human SOD1 (hSOD1) mutants become symptomatic, but the high expressed enzyme is Cu-deficient, and their intrasubunit disulfide bonds are reduced. This week, Bergemalm et al. show that these changes lead to nonphysiological mitochondrial loading of stable hSOD11 mutants, ∼100?fold higher than the endogenous mouse SOD1. In contrast, mice that expressed the unstable mutants G85R and G127X had 100- and 1000-fold lower mitochondrial hSOD1 levels, suggesting that unstable mutants do not cause disease by affecting mitochondria. The authors conclude that mitochondrial accumulation of SOD1 per se does not lead to motor neuron injury and that mitochondrial overloading may complicate interpretations of studies in mouse models expressing the stable mutants.
1. Pain Makes CaMKII Go Up and Down
Max Larsson and Jonas Broman
Aparticularly intense painful stimulus enhances the sensation evoked by subsequent noxious stimuli. This hyperalgesia may result from facilitation of sensory neurotransmission in the spinal cord, perhaps by mechanisms similar to synaptic plasticity in other pathways. In this week's Journal, Larsson and Broman used immunolabeling of calcium/calmodulin-dependent kinase II (CaMKII) to track the associated modulation of spinal cord pathways. The translocation of autophosphorylated CaMKII to the postsynaptic density makes it one marker of synaptic plasticity. The authors injected rat hindpaws with capsaicin to induce hyperalgesia. Transganglionic tracing of the capsaicin injection site marked the nonpeptidergic class of nociceptors, but not peptidergic nociceptive neurons that express substance P and calcitonin generelated peptide. Phosphorylated CaMKII more than doubled in dorsal horn synapses formed by peptidergic neurons and fell by about one-half in synapses formed by labeled, nonpeptidergic neurons. In low-threshold mechanoreceptor neurons, CaMKII expression did not change with capsaicin treatment.
2. D1, GAD, and Working Memory
Nobuhide Kobori and Pramod K. Dash
Kobori and Dash examined deficits in working memory after a mild concussive brain injury that did not cause overt neuronal loss. The authors targeted the medial prefrontal cortex (mPFC), known to be important for working memory (WM). Rats performed a delay match-to-place task, in which they escaped a water maze to a hidden platform (the location trial), were removed from the maze, and then relocated the platform (the match trial). Brain-injured rats displayed a random search pattern during the match trial indicative of impaired WM. The memory deficits were associated with neurochemical changes in mPFC, including higher levels of D1 dopamine receptors and more neurons detected by immunolabeling for GAD67, the GABA-synthesizing enzyme. GABA receptor antagonists improved WM, and remarkably a single dose of the D1 receptor antagonist SCH23390 [R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1 H-3-benzazepine hydrochloride] reduced GAD67 expression and improved WM for several days. Pass the SCH23390, please.
3. Focusing Attention on the Thalamic Reticular Nucleus
Kerry McAlonan, James Cavanaugh, and Robert H. Wurtz
In this week's Journal, McAlonan et al. explore the "attentional searchlight" hypothesis for the thalamic reticular nucleus (TRN). The TRN consists of a thin layer of GABAergic neurons that receive excitatory inputs from the lateral geniculate nucleus (LGN) and cortex and send inhibitory output back to the LGN. The authors recorded spike rates of individual visual TRN neurons in monkeys. These neurons had a high basal firing rate, providing tonic inhibition to LGN, and short latency (∼25 ms) transient responses to visual stimuli. Because the TRN receives multimodal input, the authors examined responses in which monkeys shifted their attention from auditory to visual stimuli. Spiking of TRN neurons increased with the attentional shift, but latency and duration remained the same. The authors suggest that TRN neurons facilitate visual detection and discrimination by switching visual LGN neurons from tonic to burst firing, or by surround inhibition of nonvisual LGN neurons.
4. SOD1 Mutants in and out of Mitochondria
Daniel Bergemalm, P. Andreas Jonsson, Karin S. Graffmo, Peter M. Andersen, Thomas Br?nstr?, Anna Rehnmark, and Stefan L. Marklund
CuZn superoxide dismutase (SOD1)mutations underlie some cases of familial amyotrophic lateral sclerosis (ALS). Mutations cause a cytoxic gain-of-function of this antioxidant molecule that is expressed in the cytosol as well as the mitochondrial intramembrane space. Transgenic mice expressing high levels of stable human SOD1 (hSOD1) mutants become symptomatic, but the high expressed enzyme is Cu-deficient, and their intrasubunit disulfide bonds are reduced. This week, Bergemalm et al. show that these changes lead to nonphysiological mitochondrial loading of stable hSOD11 mutants, ∼100?fold higher than the endogenous mouse SOD1. In contrast, mice that expressed the unstable mutants G85R and G127X had 100- and 1000-fold lower mitochondrial hSOD1 levels, suggesting that unstable mutants do not cause disease by affecting mitochondria. The authors conclude that mitochondrial accumulation of SOD1 per se does not lead to motor neuron injury and that mitochondrial overloading may complicate interpretations of studies in mouse models expressing the stable mutants.