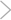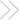In diabetic LDLR^sup -/-^ mice, an ectopic BMP2-Msx2 gene regulatory program is upregulated in association with vascular calcification. We verified the procalcific actions of aortic Msx2 expression in vivo. CMV-Msx2 transgenic (CMV-Msx2Tg^sup +^) mice expressed 3-fold higher levels of aortic Msx2 than nontransgenic littermates. On high-fat diets, CMV-Msx2Tg^sup +^ mice exhibited marked cardiovascular calcification involving aortic and coronary tunica media. This corresponded to regions of Msx2 immunoreactivity in adjacent adventitial niyofibroblasts, suggesting a potential paracrine osteogenic signal. To better understand Msx2-regulated calcification, we studied actions in 10T1/2 cells. We found that conditioned media from Msx2-transduced 10T1/2 cells (Msx2-CM) is both pro-osteogenic and adipostatic; these features are characteristic of Wnt signaling. Msx2-CM stimulated Wnt-dependent TCF/LEF transcription, and Msx2-transduced cells exhibited increased nuclear β-catenin localization with concomitant alkaline phosphatase induction. Msx2 upregulated Wnt3a and Wnt7a but downregulated expression of the canonical inhibitor Dkk1. Dkk1 treatment reversed osteogenic and adipostatic actions of Msx2. Teriparatide, a PTH1R agonist that inhibits murine vascular calcification, suppressed vascular BMP2-Msx2-Wnt signaling. Analyses of CM V-Msx2Tg^sup +^ mice confirmed that Msx2 suppresses aortic Dkk1 and upregulates vascular Wnts; moreover, TOPGAL^sup +^ (Wnt reporter); CMV-Msx2Tg^sup +^ mice exhibited augmented aortic LacZ expression. Thus, Msx2-expressing cells elaborated an osteogenic milieu that promotes vascular calcification in part via paracrine Wnt signals.
Nonstandard abbreviations used: ALP, alkaline phosphatase; BMP, bone morpbogenetic protein; CM, conditioned media; CMV, cycomegalovirus; CMV-Msx2Tg^sup +^, CMV-Msx2 transgenic; CVC, calcifying vascular cell; Dkk1, dickkopf homologue 1; EGFP, enhanced green fluorescent protein; ESRD, end-stage renal disease; LacZ, bacterial β-galactosidase; LDLR, LDL receptor; LEF, lymphoid enhancer factor; LRP, LDL receptor-related protein; Osx, osterix; PTH, parathyroid hormone; PTH1R, parathyroid hormone type 1 receptor; RSVLUC, Rous sarcoma virus minimal promoter LUC; SOST, sclerostin; TCF, T cell factor; TOPFLASH, TCF/LEF optimal promoterLUC reporter; TOPGAL, TCF/LEF1 optimal promoter- galactosidase reporter.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 115:1210-1220(2005). doi:10.1172/JCI200524140.
Introduction
Vascular calcification is a common feature of aging, diabetes, dyslipidemia, abnormal valve biomechanics, and end-stage renal disease (ESRD) (1-3). Four histoanatomic variants - medial artery calcification, atherosclerotic intimai calcification, valvular calcification, and soft tissue calciphylaxis - can be readily identified (1-3). While calciphylaxis results from passive tissue mineralization arising from thermodynamic excess of the calcium phosphate product, the first 3 variants proceed in part via the matrix vesicle-mediated calcification characteristic of intramembranous and endochondral bone formation (1-3). Once macrovascular calcification was considered benign, but its deleterious consequences have now become clear (1-3). Medial artery calcification presages a 4-fold increased risk for lower extremity amputation and 2-fold enhanced cardiovascular mortality in patients with type II diabetes (4). Medial artery calcification and atherosclerotic intimal calcification independently portend mortality in patients with ESRD (5). Aortic arch calcification correlates with stroke risk, and aortic valve calcification is a strong predictor of disease progression in patients with asymptomatic aortic stenosis (reviewed in refs. 1, 2). In medial, intimal, and valvular calcification, immune responses initiated by oxidized LDL (6) contribute to low-grade vascular inflammation with upregulation of TNF-α (7) and bone morphogenetic protein 2 (BMP2) (8). BMP2 expression is activated by TNF-α (9), oxidized lipids (9), and hyperglycemia (7, 10) in vascular myofibroblasts and endothelium. BMP2 is necessary for osteogenic differentiation of mesenchymal cells (11); via Smaddependent signals, BMP2 upregulates osteogenic (e.g., Msx2, Dlx5, Osterix [Osx]) and chondro-osteogenic (Runx2/Cbfa1, Sox9) transcription factors in mineralizing mesenchymal progenitors (12). Demer first demonstrated BMP2 expression as a feature of atherosclerotic calcification (8). Following these seminal observations, others detailed the temporospatial expression of BMP2-regulated intramembranous and endochondral gene expression programs in calcific vasculopathy (13, 14). Thus, in addition to being key inhibitors of tissue mineralization (15-18), osteogenic programs actively contribute to vascular calcification.
We previously described high-fat diets as promoting vascular calcification and upregulating aortic BMP2 and Msx2 expression in LDL receptor-null (LDLR^sup -/-^) mice (10, 14, 19). In response to high-fat diets, male LDLR^sup -/-^ mice develop diabetes and dyslipidemia (14). In this model of disease initiation, in situ hybridization studies demonstrated that the pattern of Msx2 expression in cardiac valves and adventitial myofibroblasts overlaps the expression pattern of Acta2 (i.e., VSMC α-actin) (14). Msx2 is a BMP2-inducible transcription factor that controls craniofacial mineralization (20). While promoting early osteoblast development, Msx2 inhibits osteoblast terminal differentiation; Msx2 is downregulated by inhibitory protein-protein interactions with Dlx family members that trigger full elaboration of the osteoblast phenotype (21). We recently demonstrated that Msx2 programs osteogenic differentiation of vascular myofibroblasts (10). This occurs in part via cell-autonomous actions that diminish PPARγ expression (10). We now extend these observations by showing that cells that express Msx2 also elaborate paracrine signals that promote osteogenic differentiation and suppress adipogenesis. Msx2 upregulates expression of multiple Wnt ligands (22), downregulates the LDL receptor-related proteins 5 and 6 (LRP5/6) antagonist dickkopf homologue 1 (Dkk1) (23), and enhances aortic canonical Wnt signaling (23). Therefore, Msx2 activates nuclear localization of β-catenin, a transcriptional coadapter of T cell factor/ lymphoid enhancer factor (TCF/LEF) indispensable for osteoblast differentiation and osteogenic calcification (24).
Figure 1
Enhanced cardiovascular calcification in CMV-Msx2Tg+ mice. Mice transgenic for Msx2 expression were generated as described in Methods. A total of 12 male siblings (6 CMV-Msx2Tg^sup +^, 6 nontransgenic, C57BL/6 background) were placed on high-fat diets for 4 months, sacrificed, and analyzed for cardiovascular calcification by alizarin red histochemistry. Five of 6 CMV-Msx2Tg^sup +^ mice exhibited cardiovascular calcium deposition (A), while none of the nontransgenic wild-type siblings were significantly afflicted (B). Note the pronounced calcium staining in the tunica media of the left coronary artery (LCA) and aorta, the right ventricular myocardium, and the pulmonary outflow tract. Magnification, 100. (C) CMV- Msx2Tg^sup +^. Higher-power magnification of panel A, 400. Note marked calcification associated with cells of the tunica media (arrows) and the occasional much weaker and diffuse staining in the adventitia (arrowhead).
Results
CMV-Msx2 transgenic mice exhibit cardiovascular calcification in areas overlapping and adjacent to Msx2 expression. High-fat diets upregulate the vascular expression of BMP2 and Msx2 (10, 14, 19), signaling molecules that participate in osteoblast differentiation and intramembranous ossification (12, 20). Since high-fat diets promote vascular calcification in male LDLR^sup -/-^ mice (14, 25) and BMP2-Msx2 signaling promotes osteogenic mineralization of cultured myofibroblasts (10), we proposed that Msx2 exerts procalcific actions during macrovascular calcification in vivo. To test this notion, we generated CMV-Msx2Tg^sup +^ (cytomegalovirus immediate early promoter-Msx2 transgenic) mice, a model previously demonstrated to recapitulate features of Msx2 action during craniosynostosis (26). We examined effects in LDLR^sup +^ C57B1/6 mice, a background of intermediate susceptibility to diet-induced vascular disease as compared with LDLR^sup -/-^ mice (27). Sixteen weeks of high-fat diet had little effect on vascular calcification in nontransgenic mice (Figure 1B). In contrast, in 5 of 6 male CMV- Msx2Tg^sup +^ siblings, pronounced cardiovascular calcification, revealed by alizarin red calcium staining, was observed (Figure 1A). Dense calcium deposition was observed in the aortic media, coronary arteries, pulmonary outflow tract, and adjacent right-ventricular myocardium (Figure 1A). In transgenic mice, the CMV promoter drives cell-type selective expression, directing expression in tissues subject to high levels of CMV pathology (28). In the vasculature, endothelial cells and vascular smooth muscle cells support transgenic CMV promoter activity (28); RT-PCR demonstrates that aortic expression of Msx2 is enhanced 3-fold in CMV-Msx2Tg^sup +^ animals, thus confirming cardiovascular expression of the Msx2 transgene (see below).
To further characterize cardiovascular expression of Msx2, we performed immunohistochemistry for Msx2 protein. Msx2 expression is most intense in a subpopulation of adventitial cells (Figure 2A) and at sites of cardiovascular valve leaflet insertion (Figure 2B). Moreover, patchy foci of right ventricular interstitial cardiac myofibroblasts also expressed Msx2 (not shown). Intriguingly, the majority of aortic medial cells - VSMCs - lacked significant Msx2 expression even though medial calcium deposition was widely distributed (Figures 1 and 2). We next examined expression of alkaline phosphatase (ALP), a key osteogenic enzyme upregulated by Msx2 in cell culture (10) and required for osteogenic mineralization (18). As shown in Figure 2C, ALP expression also localized to cells occupying the medial compartment in aorta and coronary arteries - the sites of vascular calcification. To confirm induction of an osteogenic-like gene regulatory process, we studied expression of sclerostin (SOST), an osteocyte protein (29), by Western blot. SOST is increased in the aortas of CMV-Msx2Tg^sup +^ mice compared with nontransgenic siblings (Figure 2D); no differences were observed in the abundance of β-actin. Of note, no differences in serum calcium (8.27 0.43 mg/dl vs. 8.25 0.53 mg/dl, P = 0.94), delipidated serum phosphorus (2.17 0.37 mg/dl vs. 2.44 0.17 mg/ dl) (P = 0.52), or aqueous calcium phosphate product (18.1 3.4 mg^sup 2^/dl^sup 2^ vs. 20.1 1.5 mg^sup 2^/dl^sup 2^, P = 0.60) were observed between wild-type and transgenic siblings, respectively. Thus, augmented aortic Msx2 expression provides signals that enhance cardiovascular calcification.
Conditioned media from Msx2-transduced cells enhanced ALP induction and inhibited lipid accumulation in 10T1/2 mesenchymal cells. Because areas of medial calcification are adjacent to regions of greatest adventitial Msx2 expression, we hypothesized that Msx2- expressing myofibroblasts might elaborate pro-osteogenic signals that function in a paracrine fashion. We previously demonstrated that the C3H10T1/2 cell system represents a highly useful cell culture model for studying effects of Msx2 on osteogenic, adipogenic, and smooth muscle differentiation (10). This mesenchymal progenitor shares features of vascular myofibroblasts and can readily adopt the mural VSMC fate when transplanted in vivo (30). Therefore, we assessed whether the conditioned media (CM) from Msx2- transduced 10T1/2 cells could alter osteogenic and/or adipogenic differentiation. As compared with CM from SFG-bacterial β- galactosidase (SFG-LacZ) controls, CM collected from 10T1/2 cells transduced with SFG-Msx2 (Msx2) significantly augmented ALP activity in heterologous 10T1/2 cells (Figure 3A). An even greater effect was observed in adipogenesis assays; CM from SFG-Msx2 cells markedly inhibited oil red O lipid accumulation in 10T1/2 cells cultured under adipogenic conditions (Figure 3B). To confirm that Msx2- expressing cells elaborate paracrine signals, we transiently expressed either enhanced green fluorescent protein (EGFP) or EGFP- Msx2 fusion protein in 10T1/2 cells, then imaged GFP and ALP activity (Vector Red) by dual immunofluorescence in cultured regions of low transfection efficiency. As compared with EGFP controls, 10T1/ 2 cells adjacent to cells expressing EGFP-Msx2 stain more intensely for ALP (Figure 3C). Thus, cells expressing Msx2 upregulate paracrine signals that control mesenchymal cell differentiation.
Figure 2
Expression of Msx2 in adventitial cells adjacent to segments of aortic and coronary medial calcification. Msx2 expression was visualized by immunohistochemistry in cardiovascular tissues from male CMV-Msx2Tg^sup +^ mice. Background signal arising from nonspecific binding of secondary antibody was assessed in the absence of primary antibody. Note expression of Msx2 transgene in adventitial cells (A) and cells at the aortic valve insertion (B). Msx2 expression was also observed in cardiac myofibroblasts (not shown). (C) Adjacent sections of cardiovascular tissues from male CMV-Msx2Tg^sup +^ mice were analyzed for Msx2 or ALP by immunohistochemistry. Alizarin red staining was used to determine patterns of tissue calcium deposition. While Msx2 is highly expressed in adventitial cells, ALP expression and calcification are most intense in the tunica media. (D) Western blot analysis of aortic proteins extracted from Msx2Tg^sup +^ compared with those from nontransgenic siblings. The expression of the osteocyte marker SOST was greater in aortas of CMV-Msx2Tg^sup +^ mice (lanes 2, 4, 6, and 8) as compared to SOST expression observed in aortas of nontransgenic siblings (lanes 1,3,5, and 7). All animals were fed high-fat diets as outlined in Methods. Arrows in A-C indicate nuclear staining. No 1 Ab, no primary antibody added.
CM from Msx2-transduced cells enhances TOPFLASH activity, andMsx2 enhances nuclear ft-catenin accumulation. Several polypeptides, including Wnt family members (11, 31), have been shown to promote osteogenesis and inhibit adipogenesis. Since Wnts are key modulators of osteogenic mesenchymal cell fate (22, 32), we examined the effects of Msx2 CM on canonical Wnt signaling using activation of the Wilt-responsive TCF/LEF transcriptional reporter-LUC reporter, TOPFLASH (33). As compared with CM from SFG-LacZ controls (10), CM collected from 10T1/2 cells transduced with SFG-Msx2 (10) significantly augmented TOPFLASH activity (Figure 4A). Rous sarcoma virus minimal promoter LUC (RSVLUC), a minimal promoter-LUC reporter lacking TCF/LEF cognates, was not regulated by Msx2-CM or Wnt3a-CM (not shown). We cocultured TOPFLASH-transfected 10T1/2 reporter cells onto transduced SFG-LacZ and onto SFG-Msx2 cell monolayers and then compared the results. Reporter cell TOPFLASH activity was greater on SFG-Msx2 layers as compared with SFG-LacZ control cell feeder layers, indicating enhanced paracrine Wnt signaling (Figure 4B). A key component of Wnt-regulated TCF/LEF-dependent transcription is nuclear localization of the transcriptional coadapter, β-catenin (33). Thus, we studied the effect of Msx2 expression on β-catenin localization. As shown in Figure 4C, in cells expressing control SFG-LacZ virus, β-catenin is localized primarily in the cytoplasm. By contrast, SFG-Msx2 markedly enhances (by 15-fold) the nuclear localization of β-catenin (Figure 4, C and D). Thus, Msx2 enhances canonical Wnt signaling in cultures of 10T1/2 cells.
Msx2 regulation of 10T1/2 osteogenic versus adipogenic differentiation is inhibited by Dkk1. Canonical Wnt signaling is negatively regulated by Dkk1, an inhibitory ligand for the cell surface receptors LRP5 and LRP6 (11, 23). To further test the role for paracrine Wnt signaling in Msx2 actions, we studied the effects of recombinant purified Dkk1 protein (Figure 5A). As shown in Figure 5B, Dkk1 inhibits Wnt3a induction of TOPFLASH activity in 10T1/2 cells, consistent with its inhibitory actions on canonical Wnt signaling. In contrast, an irrelevant recombinant protein - Tubedown- 100 (34) - has no effect. Dkk1 has no effect on basal TOPFLASH (Figure SB) reporter activity. RSVLUC, a minimal promoter - LUC reporter lacking TCF/LEF cognates, was not regulated by Wnt3a or Dkk1 (not shown). We next examined the effects of Dkk1 on ALP induction and adipogenesis. Dkk1 (1 g/ml) inhibited Msx2-induced ALP activity in 10T1/2 cells by 50% (Figure SC). An even greater effect was observed on Msx2-regulated adipogenesis. Msx2 expression suppressed oil red O accumulation by more than 80% (Figure SD); however, Dkk1 partially reversed Msx2 inhibition, increasing oil red O accumulation 3- to 4-fold compared with vehicle-treated cells. Thus, both Msx2 enhancement of osteogenesis and Msx2 inhibition of adipogenesis are antagonized by Dkk1, indicating that Wnt-like paracrine signals contribute to Msx2 actions in 10T1/2 cells.
Figure 3
CM from Msx2-expressing 10T1/2 cells enhances ALP activity and suppresses adipogenesis. C3H10T1/2 cells were transduced with retroviruses expressing either Msx2 or LacZ, and CM was harvested. Subsequently, naive 10T1/2 cells were induced to undergo either osteogenic or adipogenic differentiation in the presence of CM from either control LacZ cells or Msx2-expressing cells. (A) CM from Msx2- expressing cells supports significantly higher levels of ALP activity than CM from control cells. Percentages indicate the percent of the total cultured cell population that stains with oil red O. (B) CM from Msx2-expressing cells suppresses adipogenesis as measured by oil red O staining. (C) 10T1/2 cells were transiently transfected with expression vectors encoding either EGFP or EGPF- Msx2 fusion protein. Four days later, cells were fixed, and dual immunofluoresence was carried out as previously described, using Vector Red to image ALP. Arrows indicate cells expressing either EGFP or EGFP-Msx2 fusion proteins (green, EGFP-Msx2 in nuclear compartment). Intense ALP staining . appears adjacent to cells expressing EGFP-Msx2. *P < 0.01
Msx2 suppresses Dkk1 expression and enhances Wnt3a and Wnt7a expression in primary aortic myofibroblasts and 10T1/2 cells. During development, vascular myofibroblasts and mesodermal cells express high levels of Wnt ligands, including Wnt2, Wnt3, Wnt5, and Wnt7 (22). Moreover, Dkk1, LRPS, and LRP6 have been shown to be expressed in VSMCs (22). Therefore, we examined whether transduction of 10T1/ 2 cells with SFG-Msx2 altered expression profiles of Wnt ligands, the prototypic antagonist Dkk1, or its receptor targets LRPS and LRP6. In triplicate, primary aortic myofibroblasts or 10T1/2 cells were transduced either with SFG-LacZ control or SFG-Msx2 virus; RNA was then isolated and gene expression quantified by fluorescence RT- PCR (see Methods). Transduction of 10T1/2 or aortic cell cultures with SFG-Msx2 enhances Msx2 mRNA accumulation approximately 5,000- fold as compared with that in SFG-LacZ control cells (not shown). In both cell backgrounds, Msx2 suppressed Dkk1 expression by more than 80%, while Wnt3a, Wnt5b, and Wnt7a were upregulated 3- to 9-fold (Figure 6). The Wnt7a response in primary aortic myofibroblasts was much greater, induced 192 6.3 fold (P < 0.001) by Msx2 expression (not shown). In primary cells, no significant changes were observed in the expression of the multifunctional ligand Dkk2, Wnt2, or the Wnt coreceptor LRP6. As shown in Figure 6, LRP5 and Wnt5a were either unaffected or variably upregulated 2- to 3-fold, depending upon cell background. Thus, expression of the transcription factor Msx2 in myofibroblasts alters the gene expression profiles of Wnt- signaling regulators, upregulating key Wnt family members and suppressing the canonical antagonist Dkk1.
CMV-Msx2Tg^sup +^ mice exhibit reduced aortic Dkk1 and enhanced aortic Wnt expression compared with nontransgenic littermates. Our studies of 10T1/2 and primary aortic myofibroblast cell cultures indicate that Msx2 can upregulate expression of Wnt ligands, suppress Dkk1, and thus promote canonical Wnt signaling. To evaluate whether Msx2 regulated Wnt signaling in vivo, total aortic RNA was isolated from the aortas of either CMV-Msx2Tg^sup +^ (n = 5) or wild- type (n = 5) male siblings. Gene expression was quantified in independent duplicates for each aortic specimen as detailed (19). Msx2 mRNA accumulation was increased 3-fold in CMV-Msx2Tg^sup +^ as compared with nontransgenic littermates (Figure 7). Consistent with our in vitro observation, levels of aortic Dkkl expression in CMV- Msx2Tg^sup +^ were reduced by approximately 80% as compared with those in nontransgenic littermates. Similarly, the levels of the canonical Wnt agonist Wnt3a were upregulated 3-fold. Thus, augmented aortic Msx2 expression reduces expression of Dkk1 while upregulating expression of specific Wnt ligands.
Human parathyroid hormone (PTH^sub 1-34^), a parathyroid hormone type 1 receptor (PTH1R) agonist that suppresses vascular calcification in vitro, downregulates vascular mesenchymal Msx2-Wnt signaling. In addition, PTH1R agonists have been shown to inhibit VSMC calcification in vitro (35), and we recently demonstrated that 1 such agonist, teriparatide, also known as PTH^sub 1-34^, suppresses cardiovascular calcification in vivo (19). PTH^sub 1-34^ was shown to suppress aortic Msx2 mRNA accumulation in vivo and Msx2 expression in vitro in cultured aortic adventitial myofibroblasts (19). Therefore, to further establish the relationships between aortic Msx2-Wnt signaling and vascular calcification, we examined the effects of PTH^sub 1-34^ on the dynamics of vascular Msx2-Wnt signaling. As shown in Figure 8, PTH^sub 1-34^ treatment concomitantly suppressed Msx2, Wnt3a, and Wnt7a expression in aortic tissues (Figure 8A). PTH^sub 1-34^ had little if any effect on aortic BMP2 mRNA accumulation. PTH^sub 1-34^ actions occurred in part via direct actions on the myofibroblast PTH1R; in cultured aortic myofibroblasts, teriparatide inhibited BMP2-augmented expression of Msx2, Wnt3a, and Wnt7a (Figure 8B). Consistent with this, PTH^sub 1-34^ suppressed BMP2 activation of Smad-dependent transcription from a multimerized Smad cognate (Figure 8C). Furthermore, PTH^sub 1-34^ inhibited Wnt3a activation of the multifunctional Msx2 enhancer (36) (Figure 8D) but had little effect on basal transcription from these reporters (Figure 8 and data not shown). Thus, PTH^sub 1-34^, a PTH1R agonist that suppresses vascular calcification (19), concomitantly suppresses Msx2 induction and expression of vascular canonical Wnts that are targets of Msx2.
Figure 4
Regulation of TCF/LEF1 activity and β-catenin by Msx2 in 10T1/2 cells. (A) 10T1/2 cells were transfected with the TOPFLASH reporter and treated with either CM from control cells expressing LacZ or CM from cells expressing Msx2. Cell extracts were analyzed for LUC reporter activity. Cells treated with CM from Msx2- transduced cells supported higher levels of TOPFLASH activity than cells treated with control CM. *P < 0.05. (B) 10T1/2 cells were transiently transfected with TOPLASH and harvested 1 day layer; these reporter cells (57,000 cells per well) parachuted onto feed layers of SFG-LacZ or SFG-Msx2 cell cultures (20,000 cells per well) and were harvested for determination of LUC reporter activity 24 hours later. SFG-Msx2 feeder layers supported higher levels of TOPFLASH activity than SFG-LacZ controls. "P < 0.001. (C) Subcellular localization of (3-catenin in control (LacZ) cells compared with that in cells expressing Msx2. While excluded from the nucleus in control cells, β-catenin accumulates in the nucleus of cultured cells expressing Msx2. (D) As quantified from digital images, the percentage of cells staining for nuclear β-catenin is enhanced approximately 15-fold by Msx2.
Figure 5
Dkk1 suppresses TCF/LEF activity, inhibits Msx2-induced ALP, and partially reverses Msx2 suppression of adipogenesis. Recombinant purified Dkk1 (A, thick arrow) suppresses Wnt3a activation of the TOPLASH reporter (B). Recombinant Tubedown-100 (Tbdn100) protein (A, thin arrow) is a negative control (B). Std, protein molecular weight standard. (C) Dkk1 inhibits Msx2-enhanced ALP activity. (D) Dkk1 reverses Msx2 inhibition of adipogenesis (oil red O staining). LacZ, CM from SFG-LacZ-transduced cells. Msx2, CM from SFG-Msx2 transduced cells. *P < 0.05 for comparisons indicated by arrows in C and D.
TCF/LEF1 optimal promoter-galactosidase reporter mice transgenic for CMV-Msx2 (TOPGAL^sup +^;CMV-Msx2Tg^sup +^ mice) exhibit higher levels of vascular LacZ reporter expression as compared with nontransgenic littermates. We wished to evaluate whether augmented aortic Msx2 expression enhances canonical Wnt signaling in vivo. Thus, we studied effects of the Msx2 transgene on Wnt-dependent TCF/ LEF1 transcription using TOPGAL mice (37). In TOPGAL mice, expression of LacZ from a LEF-driven transgene is entrained to TCF/ LEF1-dependent transcription. CMV-Msx2Tg^sup +^ mice were crossed with TOPGAL mice and aortic LacZ expression compared between CMV- Msx2Tg^sup +^;TOPGAL and nontransgenic TOPGAL siblings. TOPGAL^sup +^Msx2^sup +^ mice express higher levels (P = 0.04, n = 5-7 animals per group) of LacZ tnRNA in the aorta as compared with TOPGAL^sup +^ mice (Figure 9). No augmentation of GAPDH mRNA accumulation was observed. Histochemistry for aortic LacZ confirmed that TCF/LEF signaling was activated in the medial compartments undergoing calcification (Figure 10). Thus, the LacZ reporter is augmented in TOPGAL^sup +^Msx2^sup +^ compared with TOPGAL^sup +^ control siblings, indicating enhanced aortic Wnt signaling in response to Msx2 expression.
Figure 6
Msx2 suppresses Dkk1 but induces Wnt3a and Wnt7a mRNA. RNA was extracted from aortic myofibroblasts (A) or 10T1/2 cells (B) transduced with virus expressing either LacZ (control) or Msx2 and mRNA for LRP5/6 ligands quantified by fluorescence RT-PCR. Data are expressed as the relative mRNA accumulation in cells expressing Msx2 compared with that in cells expressing LacZ (18S normalized). Basal levels of mRNA accumulation were similar in naive and SFGLacZ control cells (not shown). Dkk1 is suppressed by Msx2, while Wnt3a, Wnt5a, Wnt5b, and Wnt7a are induced. In primary cells, Wnt7a was induced 192 6.3 fold (P < 0.001 ; not shown). Note that Msx2 had no effect on LRP5 or LRP6 mRNA in primary myofibroblasts but that it weakly induced LPP5 mRNA in 10T1/2 cells. * P < 0.05; ** P < 0.001.
Discussion
Demer and colleagues first identified the expression of the bone morphogen BMP2 by pericytic myofibroblasts in segments of human calcifying artery (8). These VSMC α-actin^sup +^ myofibroblasts form alizarin red-staining calcified nodules, which contain mineralized calcium phosphate at the stoichiometric signature of hydroxyapatite (8). We determined that both BMP2 and Msx2 are upregulated in aortic adventitial and valve myofibroblasts in LDLR^sup -/-^ mice fed high fat diets (10, 14). Our in vitro studies demonstrated that Msx2 promotes the osteogenic differentiation and mineralization of primary aortic mesenchymal progenitors and 10T1/2 cells. Msx2 upregulates ALP activity while suppressing competitive regulatory programs necessary for adipogenesis (10) - an observation confirmed by others (38). We now extend our analyses to demonstrate pro-osteogenic actions of aortic Msx2 expression in vivo. On the C57B1/6 background (LDLR^sup +^), non-transgenic mice are resistant to vascular calcification, even when placed on high-fat diets for 16 weeks. In contrast, CMV-Msx2Tg+ siblings exhibit vascular calcification in aortic tunica media, coronary artery tunica media, and ventricular myocardium of adjacent right ventricular outflow. The CMV promoter has been demonstrated to direct expression of genes in subsets of vascular myofibroblasts and endothelial cells in transgenic mice (28). In our transgenic model, we noted that the calcifying vascular cells (CVCs) of the coronary and aortic tunica media were adjacent to populations of adventitial cells expressing Msx2. This suggests that Msx2 expression in adventitia may alter the mineralizing potential of neighboring mural CVCs. Consistent with this, we showed that the CM from Msx2-expressing 10T1/2 mesenchymal cells is biologically active. We determined that Msx2 augments expression of canonical (22, 23) Wnt agonists, suppresses the antagonist Dkk1, and promotes nuclear localization of β- catenin and activation of TCF/LEF1 transcription. Since recombinant Dkkl partially reverses Msx2-dependent ALP induction and the suppression of adipogenesis, this provides independent evidence for canonical Wnt signaling in Msx2 actions. Although canonical Wnt signaling has been shown to upregulate Msx2 expression (36, 39), to our knowledge, this is the first demonstration that canonical Wnt signaling is activated by Msx2.
Figure 7
The CMV-Msx2 transgene suppresses aortic Dkk1 and enhances aortic Wnt5a expression. Total RNA was isolated from individual aortas of male CMV-Msx2Tg^sup +^ mice and nontransgenic male siblings, and relative accumulation of Msx2 and LRP5/6 ligands was quantified by fluorescence RT-PCR. Data are expressed as the relative aortic mRNA accumulation in CMV-Msx2Tg^sup +^ compared with that in non-Tg control siblings (18S normalized). As observed in vitro, Msx2 expression decreases Dkk1 expression but enhances Wnt3a mRNA accumulation. * P < 0.05.
The observation that mesenchymal cells that express Msx2 elaborate paracrine Wnt signals has important implications for the pathobiology of vascular calcification. The tunica adventitia provides microvascularization, nutrient supply, and mesenchymal progenitors to the outer half of the tunica media in large vessels (40, 41). Based upon our results, we propose that induction of Msx2 signaling - first noted in adventitial myofibroblasts (14) - results in regionally augmented canonical Wnt signaling. Since the vector of microvascular flow is from tunica adventitia to tunica media (42), signals elaborated by adventitial myofibroblasts exert influences on medial cells via paracrine Wnt signals. Of note, a migratory, Seal^sup +^ adventitial cell that contributes to cells of the tunica media in response to mural injury has been identified (40); such stem cells may become programmed in the adventitia and contribute to the CVC pool in the microenvironment of the tunica media. Indeed, surgical resection of the adventitia markedly reduces segmentai medial artery calcification in a rat model of calcific vasculopathy (43). Such functional spatial relationships would help explain the circumferential pattern of medial artery calcification prevalent in diabetes. Adventitial genomic responses to diabetes and dyslipidemia, recently characterized as arising in response to low- grade oxidative stress and inflammation (7), have been known for decades (44). In our models, high-fat diets are required to robustly elicit calcification in genetically susceptible animals - even those with the Msx2 transgene (data not shown); future studies will assess whether it is the adventitial inflammation and oxidative stressors of diabetes that augment adventitial and medial Wnt-signaling cascades.
Figure 8
Teriparatide, a PTH1R agonist that suppresses vascular calcification, myofibroblast Wnt expression, and BMP2-Msx2-Wnt signaling. (A) Aortic mRNA levels for Msx2, Wnt3a, and Wnt7a were suppressed in diabetic dyslipidemic animals treated for 1 month with daily teriparatide (i.e., PTH^sub 1-34^). PTH^sub 1-34^ treatment suppresses cardiovascular calcification in this model (19). (B) mRNA levels for Msx2, Wnt3a, and Wnt7a are suppressed in cultured primary aortic myofibroblasts treated with PTH^sub 1-34^. (C) Regulation of SMAD-LUC reporter activity by 100 ng/ml BMP2 and 200 nM PTH^sub 1- 34^ in A7r5 cells. (D) Regulation of the Msx2-enhancer activity by Wnt3a CM (50% v/v) and PTH^sub 1-34^ in C2C12 cells. The Wnt5a- and BMP2-responsive Msx2 enhancer fragment -4104 to -3143 was cloned upstream of the RSVLUC minimal promoter-reporter. Note that PTH^sub 1-34^ inhibits Wnt5a activation of the Msx2 enhancer. * P = 0.02; ** P = 0.006.
The observation that Msx2-Wnt signaling is suppressed by teriparatide not only confirms the functional relationship between Msx2 and Wnt (Figure 11) but also provides insights into the endocrine pharmacology of vascular calcification. Intermittent teriparatide suppresses cardiovascular calcification and aortic myofibroblast Msx2 expression (19). Consistent with our data showing that Wnt3a and Wnt7a ace vascular targets of Msx2 proosteogenic actions, teriparatide suppresses aortic expression of these genes; BMP2, the bone morphogen upstream of Msx2 in this osteogenic signaling cascade (10, 45), is not affected by teriparatide. This suggests that PTH^sub 1-34^ inhibits aspects of BMP2 signaling in vascular myofibroblasts. Indeed, PTH^sub 1-34^ inhibits BMP2- induced expression of Msx2, Wnt3a, and Wnt7a. Moreover, PTH^sub 1- 34^ inhibits transcriptional activation by both BMP2 and Wnt3a. Like PTH^sub 1-34^, parathyroid hormone-related protein (PTHrP) also signals primarily through the PTH1R receptor in vascular smooth muscle cells, but via paracrine actions (35). PTHrP suppresses ALP expression and calcium deposition in VSMCs (35) and controls neointima formation (46). Conversely, the PTH1R antagonist PTHrPy- J4 enhances ALP expression and calcium deposition in vitro (35). ESRD is associated with profound changes in PTH1R signaling, with skeletal resistance to PTH1R activation and secondary hyperparathyroidism contributing to major abnormalities in systemic calcium metabolism (47). Antagonistic fragments of PTH such as PTH^sub 7-84^ accumulate in ESRD; these fragments bind and downregulate cell-surface PTH1R without signaling (48), thus inducing tissue resistance to both endocrine (PTH) and paracrine (PTHrP) signaling via this receptor or other C-terminal domain receptors (49). Thus, if our working model holds (Figure 11), the pathobiology of medial artery calcification in ESRD may be accelerated by hyperparathyroidism-induced tachyphylaxis to vascular PTH1R signaling - an endocrinopathy that subsequently unleashes pro- osteogenic vascular injury responses. Future studies will directly test this notion.
Figure 9
LacZ mRNA accumulates to higher levels in aortas of TOPGAL^sup +^;CMV-Msx2Tg^sup +^ than in those of TOPGAL^sup +^ mice. Aortic LacZ mRNA accumulation (fluorescence RT-PCR) was compared in doubly transgenic mice (TOPGAL^sup +^;CMV-Msx2Tg^sup +^) and TOGPAL^sup +^ controls. TOPGAL^sup +^Msx2^sup +^ mice accumulate higher levels of aortic LacZ reporter mRNA as compared with TOPGAL^sup +^ control littermates (P = 0.04; n = 5-7 per group), indicating augmented aortic Wnt and TCF/LEF signaling in response to the Msx2 transgene. * P = 0.04.
Figure 10
LacZ mRNA accumulates to higher levels in aortas of TOPGAL^sup +^;CMV-Msx2Tg^sup +^ than in those of TOPGAL^sup +^ mice. Aortic frozen sections were prepared from TOPGAL^sup +^;Msx2^sup +^ mice (A); TOPGAL^sup +^;Msx2- siblings (B), and wild-type mice (C); these were histochemically stained for LacZ (37). Two animals of each genotype were studied. Note the greater number of medial cells elaborating the LacZ reporter in CMV-Msx2Tg^sup +^ (A) than in nontrangenic TOPGAL mice (B), indicating augmented medial aortic Wnt:β-catenin signaling in response to the Msx2 transgene. LacZ was not observed in the tunica adventitia (A and D) but was very intense in the valvular fibrosus (D, TOPGAL^sup +^; CMV-Msx2Tg^sup +^). (A-C) Magnification, x400; nuclear fast red counterstain. (D) Magnification, x100; no counterstain. Arrows indicate tunica media cells expressing the LacZ reporter as revealed by histochemical staining (blue).
Our study has several important limitations. First, we used the CMV promoter to enhance aortic Msx2 expression in vivo. CMVMsx2Tg^sup +^ mice have been previously validated for the study of gain-of-function features of Msx2 relevant to ectopic calvarial suture mineralization (26) and represented a logical choice for our initial studies. Unlike the relatively nonselective transcriptional activity exhibited following transient transfection, the CMV immediate early promoter exhibits cell-type specificity in transgenic mice (28). Using the LacZ reporter, Baskar et al. (28) demonstrated that, although the CMV promoter drives expression in multiple organs, expression is restricted to specific cell types in each organ, including endothelial cells and certain VSMCs (28). We detected Msx2 immunoreactivity primarily in aortic adventitial cells and cardiac myofibroblasts (10, 14). Using transduced aortic adventitial myofibroblasts (VSMC α-actin^sup +^), we've demonstrated that these cells can and do contribute to Msx2- dependent actions (10). However, the broader specificity of the CMV promoter precludes us from unambiguously identifying a single cell type as the source of all Msx2-dependent signaling. In addition to pericytic myofibroblasts, microvascular endothelial cells may contribute to responses. second, the origins of vascular osteogenic progenitors have yet to be precisely mapped. Detailed analysis is currently limited in part because validated stage-specific markers of the myofibroblast lineage are lacking. Our ex vivo data coupled with our previous in situ hybridization studies indicate that adventitial myofibroblasts contribute to calcification responses (10, 14, 19). Since adventitial myofibroblasts are migratory (40), cells mobilized via proliferative expansion of the tunica adventitia may robustly calcify in the environment of the tunica media. However, Demer and colleagues have identified a subpopulation of resident cells in the tunica media - the CVC - that express pericytic myofibroblast markers, elaborate ALP, and deposit minerals in response to oxidized sterols and monocyte inflammatory signals (2, 6, 50,51). Since CVCs represent 10-30% of the medial cell population (50), the relative contribution of media versus adventitial osteogenic progenitors to mural calcification has yet to be established. The adventitial Sca1^sup +^ cell may represent the postnatal progenitor to all mural myofibroblast cell types (52). The patchy ventricular myofibroblast expression of the CMV-Msx2 transgene may recruit similar calcifying progenitors. In our models, high-fat diets are necessary to induce robust vascular calcification (ref. 14 and data not shown); thus, some diet-regulated metabolic parameter, potentially a phospholipid or oxidized sterol (6, 8, 51), synergizes with Msx2-Wnt signaling to drive medial calcification. Of note, surgical resection of the advencitia reduces segmentai medial artery calcification (43); whether this arises from lossof adventitial pro-osteogenic paracrine signals, reductions in the vascular osteoprogenitor pool, or a combination of both is unknown. Cell surface markers that stratify vascular myofibroblast stages of development are currently lacking, but will be necessary to identify precisely how Msx2-Wnt signaling controls the fate of vascular mesenchymal cells. Finally, the specific Wnt milieu regulating vascular calcification is unknown; 19 murine Wnt genes exist. We noted that both canonical agonists (Wnt5a, Wnt7) and noncanonical ligands (Wnt5) that inhibit canonical Wnt actions were upregulated by Msx2. The induction of ALP enzyme activity, a target of canonical Wnt signaling (11), is a key event, since ALP hydrolyzes organic phosphates to generate inorganic phosphate for mineral deposition; this includes pyrophosphate, a mineralization inhibitor (18, 53). The relative contribution of canonical and noncanonical Wilts to vascular disease processes has yet to be explored. Since the adventitia only very weakly stains for calcium with alizarin red, signals must exist that inhibit ALP induction and mineralization in this compartment; Wnt5 family members (54) and ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPPl) (18, 55, 56) represent attractive candidates. Future studies will relate spatial and temporal patterns of vascular ENPP expression to ALP as regulated by Msx2 and Wnt signaling.
Figure 11
Working model of Msx2 actions during vascular calcification. High- fat diets upregulate aortic BMP2 expression (10). In addition to providing progenitors for medial disease processes (40, 41, 43), Msx2-expressing adventitial myofibroblasts produce an osteogenic milieu via induction of Wnt agonists and inhibition of the LRP5/6 antagonist, Dkk1. This causes nuclear localization and activation of β-catenin - an indispensable coregulator of osteoblast differentiation and mineralization (24); as reflected by TCF/LEF- dependent transcription, activation occurs in a subset of medial CVCs. CVCs of Demer (50), perhaps derived from migratory adventitial progenitors (10, 40, 41, 43), respond by upregulating ALP expression via key osteogenic transcription factors (11). Vascular PTH1R signaling dampens Msx2-Wnt signaling. In ESRD, PTH1R downregulation (desensitization) arising from hyperparathyroidism may accelerate vascular disease. Although Osx is upregulated by Msx2 (10), the relationship between β-catenin:TCF/LEF signaling (24) and Osx (60) in mineralization has yet to be clarified. The pyrophosphate- generating (PPi-generating) enzyme ENPP1 inhibits arterial calcification, stabilizing the VSMC phenotype (55).
Methods
Materials. TOPFLASH (contains tandem TCF/LEF1 responsive elements upstream of the LUC reporter) was provided by Fanxin Long (Washington University, St. Louis, Missouri, USA). SMAD-LUC (contains tandem Smad responsive elements upstream of the LUC reporter) was provided by Xu Cao (University of Alabama, Birmingham, Alabama, USA). Wnt5a containing CM (Wnt3a-CM) was prepared from L cells that express Wnt3a (ATCC; CRL-2647); CM from wild-type L cells was used as a negative control. Rabbit anti-β-catenin was from Cell Signaling Technology. Goat anti-human Dkkl (no. AF1096) and goat anti-SOST (no. AF1S89) were obtained from R&D Systems. Goat anti-Msx2 (sc-17729), goat anti-ALP (tissue nonspecific ALP; sc- 23430), and mouse anti-β-actin (sc-8432) were purchased from Santa Cruz Biotechnology Inc. Other reagents were from Fisher Scientific and Sigma-Aldrich.
Transduction, transfection, and assays of lineage allocation and canonical Wnt signalingin 10T1/2 mesenchymalcells. Transduction of 10T1/2 cells by SFG-Msx2 retrovirus was performed as described (10). Cells transduced with SFG-LacZ were routinely used as negative controls. CM from SFG-LacZ-or SFG-Msx2-transduced 10T1/2 cells was harvested every 3 days for 10 days from confluent cultures and stored at -80C until use. For osteogenesis assays, confluent 10T1/2 cells were treated every 2 to 3 days with CM diluted with culture medium (10% FBS in Eagle's basal medium) to 50% or 80% strength (vol/ vol). ALP activity was measured 9 days later as described (10). For adipogenesis, confluent cells were treated with the indicated CM containing 0.1 M dexamethasone, 50 M indomethacin, and 5 g/ml insulin for 13 days. Adipocyte number was counted after oil red 0 staining (10). As indicated, 1 g/ml recombinant purified Dkk1 protein (see below) was added to culture conditions. To detect activity of canonical Wnts in CM, 10T1/2 cells were transfected with TOPFLASH; 6 hours later, cells were treated with 70% (vol/vol) LacZ- CM or Msx2-CM for 48 hours and LUC reporter activity measured as described (10). The eukaryotic expression vector for EGFP-Msx2 fusion protein was constructed in pEGFP-C1 (BD Biosciences).
Real-time fluorescence RT-PCR. To measure relative mRNA levels of Wnt/Dkk1-signaling molecules, fluorescence RT-PCR was performed as detailed (19). Validated amplimers were: Msx2, 5'- ACCACGTCCCAGCTTCTAGC-3' and 5'-GCTCTGCGATGGAGA-GGTACTG-3'; Wnt3a, 5'- CATGCACCTCAAGTGCAAATG-3' and 5'-TGAGGAAATCCCCGATGGT-3'; Wnt5b, 5'- TGTGGAGACAACGTGGAGTACG-3' and 5'-TGTAGGTTCATGAGAGCTCGGC-5'; Wnt7a, 5'-TGGATGCCCGGGAGATC-3' and 5'-CCGACCCGCCTCGTTATT-3'; Dkk1,5'- GCTGCATGACGCACGCTAT-3' and 5'-AGAGGGCATGCATATTCCATTT-3'; Dkk2, 5'- CACACTCCAAGATGCCTCATATAAA-3' and 5'-AAAACCCATCAATGCAGTCTGA-3'; LRP5, 5'-GAGCGAGGAGGCCATCAA-3' and 5'-GCCCGAGATGACAATGTTCTG-3'; LRP6, 5'- TGTGGGCCTGACCGTGTT-3' and 5'-TTCGAGCCTGGACCTTGGT-3'; LacZ, 5'- TGGCGTTACCCAACTTAATCG-3' and 5'-GCGGGCCTCTTCGCTATTAC-3'. Relative mRNA abundance was calculated as percentage of GAPDH or 18S RNA in each sample. Amplimers for 18S and GAPDH have been described (19).
Western hot analysis and immunoflaorescence. Cell pellets and aortic tissues were rinsed with PBS, then extracted at 4C with a hand-held Tissue-Tearor rotary homogenizer (BioSpec Products Inc.) in S volumes of 20 mM HEPES, pH 7.4, 0.5% Triton X-100, 0.3 M NaCl, 0.5 m M EDTA, 0.5 mM DTT, 1 nM okadaic acid, and 0.1 M sodium vanadate, supplemented with a comprehensive protease inhibitor cocktail. After centrifugation, 30 g of whole cell or tissue protein extract was resolved by SDS-PAGE then electotransferred to Immobilon- P membranes (Millipore Corp.); proteins were immunovisualized by Western blot (57). In subconfluent cultures, indirect immunofluorescence was used to localize β-catenin in 10T1/2 cells transduced with SFG-LacZ or SFG-Msx2 as previously detailed (57). ALP was visualized by Vector Red ALP substrate kit VC-SK-S100 as recommended by the manufacturer (Vector Laboratories).
Generation of recombinant purified Dkk1. Dkk1 coding sequence was obtained from human aorta cDNA (BD Biosciences - Clontech) using PCR to introduce 5'-NdeI and 3'-BamI sites, then cloned into pET16b (EMD Biosciences). Dkkl protein was purified following the Prockop protocol (58). SDS-PAGE, Western blot analysis, and inhibition of Wnt3a-induced TOPFLASH activation in transfected 10T1/2 cells were used to characterize Dkk1 preparations prior to implementation in osteogenesis and adipogenesis assays.
CMV-Msx2 and TOPGAL^sup +^;CMV-Msx2Tg^sup +^ mice (C57B1/6 background) were generated essentially as described (26). A single line yielding Msx2 transgene expression was analyzed. The pcDNA3- Msx2 expression construct DT21.14 (21) was digested with BglII and NruI to release the 3.2-kb fragment encoding the CMV immediate early promoter, mouse Msx2 cDNA (N-terminal Met-FLAG), bovine growth hormone 3'-UTR, and the polyadenylation signal (from pcDNA3; Invitrogen Corp.). This fragment was gel-purified with QIEX II (QIAGEN) silica resin, acetate-ethanol precipitated, resolubilized in transgene buffer (10 mM Tris pH 7.4, 0.1 mM EDTA; 20 l per 2000 ng of DNA), and transgenic mice generated via male C57Bl/6 pronuclear injection (Washington University Mouse Genetics Core). PCR genotyping for the CMV-Msx2-bGHpolyA transgene was directed toward juxtaposed Msx2 cDNA (5'-TGTGCTCCCCATCCCGCCTGTT-GGACTCTA-3') and vector 3'-UTR sequences (5'-AAGGACAGTGGGAGTGGCACCTTCCAGGGT-3') following the protocol of Stratman (59). TOPGAL mice (37) were purchased from Jackson Laboratory (genotyping amplimers: 5'- GAGTGACGGCAGTTATCTGGAAGATCAGGA-5' and 5'- GGAAACCGACATCGCAGGCTTCTGCTTCAATCA-5'). TOPGAL mice were bred with CMV-Msx2Tg^sup +^ mice to generate CMV-Msx2Tg^sup +^;TOPGAL^sup +^ animals on a mixed C57Bl/5-CD1 background. Comparisons of LacZ mRNA and activity were made between CMV-Msx2Tg^sup +^;TOPGAL^sup +^ and TOPGAL^sup +^ littermate controls.
Cardiovascular histochemistry, immunohistchemistry, and serum biochemistry. Animals were fed Picolab Rodent Diet 20 (no. 5053) until challenged, beginning at 6 weeks, with high-fat diet for 16 weeks (Harlan Teklad, TD88137) (14). Male mice were exsanguinated under anesthesia (19). Animal care and experimental use was approved by the Animal Studies Committee and Review Board of Washington University School of Medicine, St. Louis, Missouri, USA. The heart and ascending aorta were harvested and paraffin sections analyzed for calcium deposition by alizarin red histochemistry (19). Msx2 (1:50) and ALP (1:200) were immunolocalized using cognate goat anti- sera, horseradish peroxidase-conjugated secondary anti-goat antibodies (1:200), and diaminobenzidine histochemistry (56). After chloroform-methanol extraction of serum phospholipids, aqueous serum phosphorus and calcium concentrations were determined using commercially available assays purchased from Sigma-Aldrich.
Statistics. Analyses were performed using Student's unpaired t test, with data presented as the mean SEM (10, 19).
Acknowledgments
This work was supported by grants HL69229, AR43731, and AR07033 from the NIH and by the Barnes-Jewish Hospital Foundation.
1. Vattikuti, R., and Towler, D.A. 2004. Osteogemc regulation of vascular calcification: an early perspective. Am. J. Physiol. Endocrinol. Metab. 286:E686-E696.
2. Abedin, M., Tintut, Y., and Demer, L.L. 2004. Vascular calcification: mechanisms and clinical ramifications. Arterioscler. Thromb. Vase. Biol. 24:1161-1170.
3. Moe, S.M., and Chen, N.X. 2004. Pathophysiology of vascular calcification in chronic kidney disease. Circ. Res. 95:560-567.
4. Lehto, S., Niskanen, L., Suhonen, M., Ronnemaa, T., and Laakso, M. 1996. Medial artery calcification. A neglected harbinger of cardiovascular complications in non-insulin-dependent diabetes mellitus. Arterioscler. Thromb. Vase. Biol. 16:978-983.
5. London, G.M., et al. 2003. Arterial media calcification in end- stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol. Dial. Transplant. 18:1731-1740.
6. Tintut, Y., et al. 2002. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation. 105:650-655.
7. Zhang, L., et al. 2003. Diabetes-induced oxidative stress and low-grade inflammation in porcine coronary arteries. Circulation. 108:472-478.
8. Bostrom, K., et al. 1993. Bone morphogenetic protein expression in human atherosclerotic lesions. J. Clin. Invest. 91:1800-1809.
9. Cola, C., Almeida, M., Li, D., Romeo, F., and Mehta, J.L. 2004. Regulatory role of endothelium in the expression of genes affecting arterial calcification. Biocbem. Biophys. Res. Commun. 320:424-427.
10. Cheng, S.L., Shao, J.S., Charlton-Kachigian, N., Loewy, A.P., and Towler, D.A. 2003. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. J. Biol. Chem, 278:45969-45977.
11. Rawadi, G., Vayssiere, B., Dunn, F., Baron, R., and Roman- Roman, S. 2003. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. J. Bone Miner. Res. 18:1842-1853.
12. Wozney J.M., and Rosen, V. 1998. Bone morphogenetic protein and bone morphogenetic protein gene family in bone formation and repair. Clin. Orthop. 346:26-37.
13. Tyson, K.L., et al. 2003. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler. Thromb. Vase. Biol. 23:489-494.
14. Towler, D.A., Bidder, M., Latifi, T., Coleman, T., and Semenkovich, C.F. 1998. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J Biol. Chem. 273:30427-30434.
15. Schafer, C., et al. 2003. The serum protein alpha 2-Heremans- Schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J. Clin. Invest. 112:357-366. doi:10.1172/ JG1200317202.
16. Rutsch, F., et al. 2001. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Patbol 158:543-554.
17. Speer, M.Y., et al. 2002. Inactivation of the osteopontin gene enhances vascular calcification of matrix GIa protein- deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J. Exp. Med. 196:1047-1055.
18. Harmey, D., et al. 2004. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am. J. Pathol. 164:1199-1209.
19.Shao, J.S., Cheng, S.L., Charlton-Kachigian, N., Loewy, A.P., and Towler, D.A. 2003. Teriparatide [human parathyroid hormone (1- 34)] inhibits osteogenic vascular calcification in diabetic low density lipoprotein recepcor-dcficient mice. J. Biol. Chem. 278:50195-50202.
20. Satokata, I., et al. 2000. Msx2 deficiency in mice causes pleiotropic defects in bone growth and ectodermal organ formation. Nat. Genet. 24:391-395.
21. Newberry, E.P., Latifi, T., and Towler, D.A. 1998. Reciprocal regulation of osteocalcin transcription by the homeodomam proteins Msx2 and Dlx5. Biochemistry. 37:16360-16368.
22. Goodwin, A.M., and D'Amore, RA. 2002. Wnt signaling in the vasculature. Angiogenesis. 5:1-9.
23. Mao, B., et al. 2002. Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature. 417:664-667.
24. Hu, H., et al. 2005. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 132:49-60.
25. Davies, M.R., Lund, R.J., and Hruska, K.A. 2003. BMP-7 is an efficacious treatment of vascular calcification in a murine model of atherosclerosis and chronic renal failure. J. Am. Soc. Nephrol. 14:1559-1567.
26. Liu, Y.H., et al. 1995. Premature suture closure and ectopic cranial bone in mice expressing Msx2 transgenes in the developing skull. Proc. Natl. Acad. Sci. U. S.A. 92:6137-6141.
27. Schreyer, S.A., Lystig, T.C., Vick, C.M., and LeBoeuf, R.C. 2003. Mice deficient in apolipoprotein E but not LUL receptors are resistant to accelerated atherosclerosis associated with obesity. Atherosclerosis. 171:49-55.
28. Baskar, J.F., et al. 1996. The enhancer domain of the human cytomegalovirus major immediate-early promoter determines cell type- specific expression in transgenic mice. J. Virol 70:3207-3214.
29. van Bezooijen, R.L., et al. 2004. Sclerostin is an osteocyte- expressed negative regulator of bone formation, but not a classical BMP antagonist. J. Exp. Med. 199:805-814.
30. Hirschi, K.K., Rohovsky, S.A., and D'Amore, P.A. 1998. PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 141:805-814.
31. Ross, S.E., et al. 2000. Inhibition ofadipogenesis by Wnt signaling. Science. 289:950-953.
32. Boyden, L.M., et al. 2002. High bone density due to a mutation in LDL-receptor-related protein 5. N. Engl J. Med. 346:1513- 1521.
33. Kim, K., Pang, K.M., Evans, M., and Hay, E.D. 2000. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LBF-I or the involvement of major G1 cell cycle regulators. Mol. Biol. Cell. 11:3509-3523.
34. Willis, D.M., et al. 2002. Regulation of osteocalcin gene expression by a novel Ku antigen transcription factor complex. J. Biol. Chem. 277:37280-37291.
35. Jono, S., Nishizawa, Y., Shioi, A., and Morii, H. 1997. Parathyroid hormone-related peptide as a local regulator of vascular calcification. Its inhibitory action on in vitro calcification by bovine vascular smooch muscle cells. Arteriosder. Thromb. Vasc. Biol. 17:1135-1142.
36. Hussein, S.M., Duff, E.K., and Sirard, C. 2003. Smad4 and beta-catenin co-activators functionally interact with lymphoid- enhancing factor to regulate graded expression of Msx2. J. Biol. Chem. 278:48805-48814.
37. DasGupta, R., and Fuchs, E. 1999. Multiple roles for activated LKF/TCF transcription complexes during hair follicle development and differentiation. Development. 126:4557-4568.
38. Ichida, F., et al. 2004. Reciprocal roles of MSX2 in regulation of osteoblast and adipocyte differentiation J. Biol. Chem. 279:34015-34022.
39. Willert, J., Epping, M., Pollack J.R., Brown, P.O., and Nusse, R. 2002. A transcriptional response to Wnt protein in human embryonic carcinoma cells. BMC Dev. Biol. 2:8.
40. Hu, Y., et al. 2004. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE- deficient mice. J. Clin. Invest. 113:1258-1265. doi:10.1172/ JCI200419628.
41. Sartore, S., et al, 2001. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Ore. Res. 89:1111-1121.
42. Heistad, D.D., Marcus, M.L., Larsen, G.E., and Armstrong, M.L. 1981. Role of vasa vasorum in nourishment of the aortic wall, Am. J. Physiol. 240:H781-H787.
43. Biijan J., et al. 1996. Modifications induced by atherogenic diet in the capacity of the arterial wall in rats to respond to surgical insult. Atherosclerosis. 122:141-152.
44. Lehmann, R., et al. 1980. Alteration of chromatin in early experimental arteriosclerosis. Artery. 8:288-293.
45. Daluiski, A., et al. 2001. Bone morphogenetic protein-3 is a negative regulator of bone density. Nat. Genet. 27:84-88.
46. Fiaschi-Taesch, N., Takane, K.K., Masters, S., Lopez- Talavera, J.C., and Stewart, A.F. 2004. Parathyroid hormone-related protein as a regulator of pRb and the cell cycle in arterial smooth muscle. Circulation. 110:177-185.
47. Slatopolsky, E., et al. 2000. A novel mechanism for skeletal resistance in uremia. Kidney Int. 58:753-761.
48. Sneddon, W.B., et al. 2003. Activation-independent parathyroid hormone receptor internalization is regulated by NHERFl (EBP50). J. Biol. Chem. 278:43787-43796.
49. Murray, T.M., Rao, L.G., Divieti, P., and Bringhurst, F.R. 2005. Parathyroid hormone secretion and action: evidence for discrete receptors for the carboxyl-terminal region and related biological actions of carboxyl-terminal ligands. Endocr. Rev. 26:78- 113.
50. Tintut, Y., et al. 2003. Multilineage potential of cells from the artery wall. Circulation. 108:2505-2510.
51. Kha, H.T., et al. 2004. Oxysterols regulate differentiation of mesenchymal stem cells: pro-bone and anti-fat J. Bone Miner. Res. 19:830-840.
52. Owens, G.K., Kumar, M.S., and Wamhoff, B.R. 2004. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84:767-801.
53. Anderson, H.C., et al. 2004. Impaired calcification around matrix vesicles of growth plate and bone in alkaline phosphatase- deficient mice. Am. J. Pathol. 164:841-847.
54. Topol, L., et al. 2003. WnC-5a inhibits the canonical Wnt pathway by ptomoting GSK-3-independent beta-catenin degradation. J. Cell Biol. 162:899-908.
55. Johnson, K., Polewski, M., van Etten, D., and Terkeltaub, R. 2004. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/-mice. Arteriosder. Thromb. Vasc. Biol. doi:10.1161/01.ATV.0000154774.71187-f0.
56. Shao, J.S., Schepp, W., and Alpers, D.H. 1998. Expression of intrinsic factor and pepsinogen in the rat stomach identifies a subset of parietal cells. AmJ. Physiol. 274:G62-G70.
57. Sierra, O.L., Cheng, S.L., Loewy, A.P., Charlton-Kachigian, N., and Towler, D.A. 2004. MINT, the Msx2 interacting nuclear matrix target, enhances Runx2-dependent activation of the osteocalcin fibroblast growth factor response element. J. Biol. Chem. 279:32913- 32923.
58. Gregory, C.A., Singh, H., Perry, A.S., and Prockop, DJ. 2003. The Wnt signaling inhibitor dickkopf-1 is required for reentry into the cell cycle of human adult stem cells from bone marrow. J. Biol. Chem. 278:28067-28078.
59. Stratman, J.L., Barnes, W.M., and Simon, T.C. 2003. Universal PCR genotyping assay that achieves single copy sensitivity with any primer pair. Transgenic Res, 12:521-522.
60. Nakashima, K., et al. 2002. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 108:17-29.
Jian-Su Shao, Su-Li Cheng, Joyce M. Pingsterhaus, Nichole Charlton-Kachigian, Arleen P. Loewy, and Dwight A. Towler
Department of Medicine, Division of Bone and Mineral Diseases, Washington University School of Medicine, St. Louis, Missouri, USA.
Received for publication December 9, 2004, and accepted in revised form on February 22, 2005.
Address correspondence to: Dwight A. Towler, Washington University Medical Center, Campus Box 8301, 660 South Euclid Avenue, St. Louis, Missouri 63110, USA. Phone: (314) 454-7434; Fax: (314) 454-8434; E-mail: dtowler@im.wustl.edu.
Jian-Su Shao and Su-Li Cheng contributed equally to this work.
Copyright American Society for Clinical Investigation May 2005
Story from REDNOVA NEWS:
Published: 2005/05/22 03:00:52 CDT
© Rednova 2004
 1
1