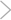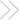Science
04-27-2001
The Mitochondrion: Is It Central to Apoptosis?
Elizabeth Finkel
Despite a tide of data suggesting that mitochondria are primary in triggering programmed cell death, a small minority of researchers argues that the primary apoptosis signals route directly to enzymes called caspases without first involving the mitochondria. The debate is hard to resolve because the events of apoptosis unfold too rapidly to determine their temporal sequence, and the use of techniques such as knocking out the genes for various components of the apoptosis pathways has provided fuel for both sides. The question is more than academic, as defects in apoptosis contribute to many major diseases; knowing exactly how to turn apoptosis on or off is therefore key to developing drugs to treat diseases in which it goes awry.
The Mitochondrion: Is It Central to Apoptosis?
Elizabeth Finkel*
Researchers studying apoptosis are divided into two camps. At issue: whether the mitochondria or enzymes called caspases are primary in triggering programmed cell death
Peer down a microscope to watch cells undergoing programmed cell death and you're in for an awe-inspiring sight. Programmed cell death--or apoptosis as it's also called--unfolds like a well-planned military operation. Within minutes, cells collapse their structural supports, digest and package their contents into membrane-bound parcels, and disappear without a trace into the bowels of scavenger cells. Because apoptosis is key to normal life--in the developing embryo it's needed to cull excess cells, for example, and later in life it eliminates damaged cells--researchers have been working feverishly to piece together the molecular circuitry that underlies this highly choreographed death program.
Within the past few years, however, the apoptosis community has found itself split into two competing camps with divergent views of just what this circuitry looks like. The dispute concerns what role the cell's mitochondria play in apoptosis.
These small, membrane-bound structures are best known as the source of the reactions producing most of the cell's energy. But a great deal of recent evidence has led many researchers to think that in most cases the mitochondria make the key decisions about whether a cell lives or dies. According to this view, when the stress signals produced by, say, lack of necessary growth factors or exposure to ultraviolet (UV) light, reach a critical threshold, the mitochondria either rupture or leak, ensuring their own demise and releasing a cocktail of factors that trigger protein-splitting enzymes called caspases. The activated caspases then rapidly cleave proteins in the cell's internal skeleton, membranes, and nucleus to bring about the characteristic hallmarks of apoptosis.
But despite a tide of data suggesting that mitochondria have their fingers on the cell's self-destruct button, a small minority of researchers argues that they are secondary agents of the cell's demise. The primary apoptosis signals, they believe, route directly to the caspases without first involving the mitochondria. Once those caspases are activated, they may target the mitochondria along with everything else in the cell, leading to further caspase activation. But these mitochondria-linked caspases merely "facilitate cellular dismemberment," says one proponent of this view, Vishva Dixit of Genentech Inc. in South San Francisco. They do not trigger it.
The debate is hard to resolve because the events of apoptosis unfold too rapidly to determine their temporal sequence, and the results from techniques such as knocking out the genes for various components of the apoptosis pathways have been inconclusive, providing fuel for both sides. "It's a funny situation," admits apoptosis researcher Pierre Golstein of the Center of Immunology of Marseille-Luminy, France. "We don't know whether the mitochondrion is at the heart of the mechanism or a lateral process. Right now I'm just counting the votes on each side."
The question of who plays the leading role in apoptosis is more than academic. Defects in apoptosis contribute to many major diseases. Too much apoptosis has been linked to nerve cell loss in conditions such as stroke and Alzheimer's disease, and too little to cancer and autoimmune disease. And knowing exactly how to turn apoptosis on or off is key to developing drugs to treat diseases in which it goes awry. "This is a very important debate," says Jerry Adams of the Walter and Eliza Hall Institute of Medical Research in Melbourne, Australia. "The central question in the apoptotic field is still unanswered. A lot hinges on that."
Center stage in apoptosis? In this view, numerous cell-death stimuli work through the mitochondrion. They cause pro-apoptotic members of the BCL-2 family, such as BAX and BAK, to either open new pores or modify existing channels in the mitochondrial membrane, releasing cytochrome c and other proteins that lead to caspase activation and cell death. BCL-2 itself, which is antiapoptotic, somehow blocks the pore or channel opening.
ILLUSTRATION: C. SLAYDEN
The mitochondrial connection
Until about 5 years ago, apoptosis researchers paid little attention to mitochondria. Early work, particularly in the roundworm Caenorhabditis elegans, did not point to their involvement, and researchers also found what appeared to be a direct pathway to caspase activation in mammalian cells. This is the Fas death receptor pathway, in which substances such as the so-called Fas ligand bind to a cell surface protein called Fas. Activation of Fas then triggers caspase-8 inside the cell.
But in 1996, a startling discovery by Xiaodong Wang's group at the University of Texas Southwestern Medical Center (UT Southwestern) in Dallas began to shift the focus to the mitochondria. Wang and his colleagues were investigating how human cells keep tabs on one of the cell's 15 or so other caspases, caspase-3, dubbed an "executioner" caspase because of the major role it plays in bringing about cell death. They found that the enzyme's activity is unleashed by cytochrome c, a mitochondrial protein then thought to be dedicated solely to energy production. "It took us a long time to believe [that finding]," recalls Sharad Kumar of the Hanson Institute in Adelaide, Australia, who studies apoptosis in the fruit fly.
Subsequent work by Wang's group showed just how cytochrome c plugs into the apoptosis circuitry. Like many other protein-degrading enzymes, the active segments of caspase-3 are buried in a "proenzyme" and need to be clipped out by another enzyme. For procaspase-3, that activation step is performed by caspase-9, and researchers found that caspase-9 requires two other proteins to make it function: Apaf-1, for apoptosis- activating factor--and cytochrome c, which floods out of the mitochondria during apoptosis. The researchers further found that known apoptosis inhibitors like the protein BCL-2 could stop that release.
Exactly how cytochrome c gets out of the mitochondrion is unclear, but 1996 work by Stephen Fesik of Abbot Laboratories in Chicago, Illinois, and his colleagues provided a clue. BCL-2 is part of a large protein family whose members divide up into opposing factions. Some, like BCL-2 or the closely related BCL-XL, block apoptosis; others, like BAX or BAK or the BH3-only proteins (so-called because they carry only the third domain of the four that characterize BCL-2 family members) are potent triggers of cell death.
Fesik and colleagues crystallized BCL-XL and found that its structure resembles that of diphtheria toxin, a protein that kills cells by punching holes in their membranes. Some researchers now think that apoptosis-triggering BCL-2 family members release cytochrome c from the mitochondria by creating pores in the mitochondrial outer membrane, while antiapoptotic members somehow preserve mitochondrial integrity.
Other researchers have proposed different exit routes for cytochrome c, ranging from existing mitochondrial pores that can be modified by pro-apoptosis BCL-2 family members to the complete rupture of the mitochondrial outer membrane. But although there is vigorous disagreement about exactly how cytochrome c gets out, the majority agrees that it is the sudden permeability of the mitochondrial outer membrane that triggers the decision to die.
Within the past year or so, that view has been buttressed by findings that cytochrome c is not the only pro-apoptotic protein released by mitochondria. Guido Kroemer's group at France's national research agency CNRS in Villejuif found that they also release three caspases and a protein known as apoptosis-inducing factor (AIF) that may play a critical role in the programmed cell death that sculpts the early embryo. And in work reported in the 7 July 2000 issue of Cell, Anne Verhagen in David Vaux's lab at the Walter and Eliza Hall Institute and Chunying Du in Wang's lab independently showed that when mitochondria release cytochrome c, they release another protein, known either as Diablo or Smac. This protein neutralizes a set of caspase inhibitors known as IAPs (for inhibitors of apoptosis), thereby freeing the caspases to do their work.
Other players make their way from their primary residence--the nucleus--to the mitochondrion in response to apoptosis signals. As reported last August by Hui Li in Xiao-kun Zhang's lab at the Burnham Institute in La Jolla, California, these include TR3, a protein previously known only as a "nuclear receptor" that regulates gene activity in response to steroid hormones. Li found that TR3 appears to quit the nucleus and relocate to mitochondria, where it causes the release of cytochrome c (Science, 18 August 2000, p. 1159). And Natalie Marchenko of the State University of New York, Stony Brook, found something similar for the p53 protein, the sentinel known to make the earliest decisions about cell life and death. It, too, was supposed to exert its effects by regulating genes, but Marchenko showed that p53 also localizes to mitochondria, where it can induce apoptosis more directly.
Even the Fas death receptor pathway, which was once thought to require only the activity of caspase-8, has recently been shown to require the help of the mitochondrial pathway. Stanley Korsmeyer's team at Washington University School of Medicine in St. Louis found that in liver cells caspase-8 cleaves a protein called BID. One of the products thus released then moves to the mitochondria, where it works through BCL family members BAK and BAX to release cytochrome c (see p. 727).
Or a side show? In the contrary view, the mitochondria come into play only secondarily, once the initiator caspases have been activated. Here Bcl-2 blocks the activity of CED-4, or its mammalian equivalent, which has not yet been found.
ILLUSTRATION: C. SLAYDEN
Another view
Despite the wealth of evidence putting the mitochondria at the center stage of cell death, a small group of researchers remains unconvinced. These include Genentech's Dixit, and Vaux and Andreas Strasser of the Walter and Eliza Hall Institute. They point out that the evidence for a primary role for the mitochondrion comes from studies of broken cells or cells grown in culture dishes. In these highly unphysiological conditions, says Vaux, "virtually everything in the Sigma [chemical company] catalog has been shown to cause apoptosis and cytochrome c release." He and the other dissenters think that more meaningful results can only come through studies in whole animals, such as C. elegans.
Researchers, particularly Robert Horvitz and his colleagues at the Massachusetts Institute of Technology (MIT) in Cambridge, have worked out the molecular events leading to apoptosis in C. elegans--and they are not triggered by permeabilizing the mitochondria. The worm's apoptosis circuitry involves just four main proteins. CED-3 (for cell death 3) is the worm's executioner caspase. It is activated by a second protein, CED-4, which is normally kept in check by the protein called CED-9. Apoptosis triggers, like the protein EGL-1, relieve that inhibition by trapping CED-9, freeing CED-4 to activate CED-3.
The worm proteins, moreover, have turned out to be similar to some identified in mammalian apoptosis. For example, CED-9 is not only structurally similar to the human apoptosis inhibitor BCL-2, but in 1993, Vaux, who was then at Stanford University, and his Stanford colleague Stuart Kim showed that the human Bcl-2 gene, when introduced into the worm, prevents the apoptosis that normally kills cells during the worm's embryonic development. That discovery led researchers to believe that the apoptosis circuitry would be similar in the two organisms, and although many dropped that idea in the face of the mitochondrial results, Vaux and a few other have remained steadfast.
They got some support about a year ago from Toshiyuki Nakagawa and colleagues at Harvard Medical School in Boston. They showed that when the cell's protein production and transport network, known as the endoplasmic reticulum, is under stress, it activates caspase-12 and triggers apoptosis with no involvement of mitochondria.
Further support comes from studies of caspase inhibitors. If activation of the enzymes is the primary event in apoptosis, inhibiting them should prevent both cell death and the mitochondrial events. And that's exactly what researchers have found with inhibitors in the fruit fly.
The final result. Mitochondria may--or may not--play a central role in triggering the apoptosis of these mouse myeloid cells (above).
CREDIT: D. VAUX/WALTER AND ELIZA HALL INSTITUTE OF MEDICAL RESEARCH
John Abrams's group at UT Southwestern showed that during the normal apoptosis of developing fly oocytes, changes occur to cytochrome c within the mitochondria that might allow it to access and activate the fly's version of the caspase activator Apaf-1. But caspase inhibitors eliminated these changes, suggesting that caspase activation is the primary event in fly apoptosis.
Furthermore, Herman Steller at MIT showed that the protein p35, a caspase inhibitor carried by certain insect viruses, prevents cell death in the retinas of flies with a condition similar to retinitis pigmentosa, a hereditary form of human blindness in which the retinal cells of the eye die abnormally by apoptosis. The flies even regained their vision. "If pressed for an answer, I would say that with respect to the main triggers of developmental apoptosis in flies, caspase action does precede events at the mitochondria," Abrams says.
Caspase inhibitors have produced much more equivocal results in mammals, however. In cultured cells exposed to various death triggers such as UV light or the chemical staurosporine, the inhibitors do stop cells from executing the typical apoptosis program. Even so, the mitochondria still release cytochrome c and the cells slowly die--a result that supports the pro-mitochondrial contingent.
The caspase camp counters this finding in two ways: The inhibitors used may not effectively block all of the cell's caspases, and cultured cells bombarded by chemicals and radiation may not behave the way cells in the living organism do. A more telling result, they argue, is the fact that caspase inhibitors like the synthetic peptide ZVAD-fmk reduce the area of tissue damage by as much as 50% in animals subjected to experimental strokes or heart attacks. "If we can keep [the neurons or heart cells] alive during the ischemic storm, they appear to be fine coming out the other end," says Don Nicholson of Merck Frosst Canada Inc. in Pointe Claire-Dorval, Quebec.
Members of the pro-mitochondrial camp maintain, however, that the problems these results pose for their point of view aren't insurmountable. Even though mitochondria may be the apoptosis initiators, they could still need caspases to finish the job. "All you need to invoke is that the caspases are providing a positive amplification loop," says Michael Hengartner, an apoptosis researcher at Cold Spring Harbor Laboratory on New York's Long Island. If so, then the mitochondria, despite having lost their cytochrome c, may be able to recover if no caspases are around. Indeed, Jean-Claude Martinou's group at the Serono Pharmaceutical Research Institute in Geneva, Switzerland, has evidence for this idea in cultured sympathetic neurons.
Attempts to clarify the issue by inactivating, or "knocking out," various genes in mice to see how that affects apoptosis have also provided fuel for both camps. Researchers have separately knocked out the genes for Apaf-1 and other proteins in the caspase 3 activation pathway. All of the resulting animals turned out to show some degree of developmentally programmed apoptosis, although it wasn't completely normal. And in work reported just 4 weeks ago in the 29 March issue of Nature, a team led by Nicholas Joza and Josef Penninger of the Amgen Research Institute in Toronto, Canada, found that whereas AIF, a mitochondrial protein, is needed for the programmed cell death that sculpts early mouse embryos, neither caspase-9 nor Apaf-1 is required. For the mitochondrial camp, this is evidence that caspases are dispensable for programmed cell death.
But the caspase proponents maintain that the residual apoptosis might simply be due to the presence of caspases other than caspase-3 or -9, including some that have not yet been identified. Strasser of the Walter and Eliza Hall Institute, in unpublished work, has evidence for such caspases. He found that in otherwise normal mice, white blood cells lacking Apaf-1 undergo normal culling during embryonic development. The same was true for white cells lacking the genes for caspase-3 or -9. "It's definitely classical apoptosis; we see all the features of caspase activation," says Strasser.
If that classical apoptosis is being brought about by alternative caspases, then they would presumably need an activator other than Apaf-1. Strasser and others propose that this could be something like the worm's CED-4. Washington University's Korsmeyer describes that as a "reasonable hypothesis," but he adds, "a number of us have done a lot of screening, looking for such a molecule, and have not found it."
Still, many gaps remain to be filled in the apoptosis circuit diagram, leaving both sides room for hope. "Having the human genome sequence in hand should speed this search [for new caspases or a CED-4 equivalent]. Time will be the best arbitrator; when the dust settles I'm confident our hypothesis will prevail," says Dixit. But so are the members of the pro-mitochondrial camp.
Indeed, given the complexity of apoptosis, which can be triggered by many different stresses acting through many different internal cell signaling pathways, it may well turn out that cells can employ different mechanisms in different circumstances. "It's not going to be black or white," predicts Hengartner. "And a good thing too; this way everyone will be right."
Elizabeth Finkel writes from Melbourne, Australia.
Copyright © 2001 by the American Association for the Advancement of Science
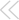
 1
1